Entomology and
Applied Science Letters
Applied Science Letters
 2020
Volume 7
Issue 4
2020
Volume 7
Issue 4

The transcription factors families (TFs) play a critical role in the response regulation of plants to abiotic stresses. The three plant lineages (E. desvauxii, C. colocynthis & Z. simplex) can be separated on the PC1 level, while R. stricta and S. Italica can be distinguished from each other on the PC2 level. A unique 225 transcripts in each of R. stricta and E. desvauxii, 85 transcripts in C. colocynthis, 92 transcripts in S. Italica, and 41 transcripts in Z. simplex were identified. All five plants were shared in only 15 transcripts. Genes had significant gene ontology (GO), R. stricta (450), E. desvauxii (475), C. colocynthis (197), S. Italica (223), and Z. simplex (75) were significantly assigned with GO terms for 29 transcriptional processes. The most abundant TF families in the five plants were MYB followed by, MYB-related, bHLH, and AP2-EREBP (1105, 803, 53 & 472 transcripts, respectively). The zinc TF families were the most transcribed and represented by 8 TF families (PLATZ, C2C2-CO-like, C3H, VOZ, C2C2-GATA, C2C2-DOF, C2H2 & ZF-HD), with the highest one (C3H TF family, 99 transcripts in both R. stricta & Z. simplex). The regulatory network showed that as heat response, 12 genes were upregulated in R. stricta and controlled several genes in some vital processes inside the plant, while E. desvauxii and S. Italica respond to heat by upregulation of 6 genes for each, C. colocynthis by regulation of 3 genes and the least one was Z. simplex by upregulation of two genes only.
Comparative Transcriptome of Transcription Factors in Rhazya Stricta and Some Other Desert Plants
Samah O. Noor1*, Dhafer A. Al-Zahrani1, Refaei M. Hussein2,3, Mohammed N Baeshen4, Tarek A. A. Moussa4,5*, Nabih A. Baeshen1, John P. Huelsenbeck6
1 Department of Biological Sciences, Faculty of Sciences, King Abdulaziz University, Jeddah, Saudi Arabia.
2 Department of Biological Sciences, Faculty of Sciences and Arts, Al Kamel Province, University of Jeddah, Saudi Arabia.
3Genetics & Cytology Dept. Genetic Engineering Division, National Research Center, Dokki, Cairo, Egypt.
4Department of Biological Sciences, Faculty of Science, University of Jeddah, Saudi Arabia.
5 Botany and Microbiology Department, Faculty of Science, Cairo University, Giza 12613, Egypt.
6 Department of Integrative Biology, University of California, Berkeley, USA.
ABSTRACT
The transcription factors families (TFs) play a critical role in the response regulation of plants to abiotic stresses. The three plant lineages (E. desvauxii, C. colocynthis & Z. simplex) can be separated on the PC1 level, while R. stricta and S. Italica can be distinguished from each other on the PC2 level. A unique 225 transcripts in each of R. stricta and E. desvauxii, 85 transcripts in C. colocynthis, 92 transcripts in S. Italica, and 41 transcripts in Z. simplex were identified. All five plants were shared in only 15 transcripts. Genes had significant gene ontology (GO), R. stricta (450), E. desvauxii (475), C. colocynthis (197), S. Italica (223), and Z. simplex (75) were significantly assigned with GO terms for 29 transcriptional processes. The most abundant TF families in the five plants were MYB followed by, MYB-related, bHLH, and AP2-EREBP (1105, 803, 53 & 472 transcripts, respectively). The zinc TF families were the most transcribed and represented by 8 TF families (PLATZ, C2C2-CO-like, C3H, VOZ, C2C2-GATA, C2C2-DOF, C2H2 & ZF-HD), with the highest one (C3H TF family, 99 transcripts in both R. stricta & Z. simplex). The regulatory network showed that as heat response, 12 genes were upregulated in R. stricta and controlled several genes in some vital processes inside the plant, while E. desvauxii and S. Italica respond to heat by upregulation of 6 genes for each, C. colocynthis by regulation of 3 genes and the least one was Z. simplex by upregulation of two genes only.
Keywords: Transcription factors, Desert plants, Heat response, Salt response, RNA sequencing.
INTRODUCTION
Plants are sessile creatures that adapt to different environmental stress conditions to establish a survival mechanism and successful reproduction. Such stressful circumstances implicate with variety of abiotic stresses such as extreme temperatures, drought, salinity, and biotic stress such as pathogen and herbivore attacks. In nature, these are generally constant or persistent. Thus, plants have an expanded approach to tolerate recurring stress and cope with it [1].
The area in which plants grow with the numerous detrimental conditions is known as “stress”. Stress is frequently defined as an extraneous factor that carries an unfavorable influence on the plant, narrows their evolution, and their opportunity of survival. The perception of stress is linked to tolerate this stress, in which plants can resist and adapt to it. Stress is mostly defined as an important variation from the optimal environmental conditions and triggers changes and responses in plant functional levels, which are reversible and might convert to permanent [2].
The feature of the state of stress in appearance is nonspecific, which exhibit primarily an expression of the severity of the disorder. Some mechanisms could consider nonspecific if it is not characterized as a pattern and nerveless of the stress type. For example, non-specific stress indications are expanded respiration, inhibition of photosynthesis, decreasing in dry matter manufacture, growth disturbance, minimizing fertility, premature senescence, leaf chlorosis, anatomical alterations, and reduced intracellular energy availability or elevated energy utilization due to repair synthesis. The responses of the cell to stress implicate modification in division and cell cycle, an adjustment in the vacuolization system, and adjustment in the cell wall structure. All these responses contribute to emphasize the stress tolerance of cells. Biochemically, plants modify different metabolism pathways to accommodate this environmental stress.
Plants transcriptional responses to these stress factors of the environment have been studied broadly over the last decades, from the transcript profiling to elucidation of the specific signaling pathways, the identification of regulatory proteins and their targets under various stress combinations. Recent researches have given rise to huge intensive information on how plants respond to different abiotic stresses such as heat, cold, drought, flooding, or salinity [3-8]. The established knowledge generated already has been used to enhance crop adaptability, e.g. over stress-inducible up-regulation of transgenes encoding enzymes that aid to produce stress garrison or stress regulators [9]. Recently scientists have become increasingly attentive to the fact that transcriptional regulation cannot be fully comprehended except if we consider the essential structure in which it takes place. The recent study demonstrates the transcription profile of the five plants (Rhazya stricta, Enneapogon desvauxii, Citrullus colocynthis, Senna Italica, & Zygophyllum simplex) which grow in adverse conditions like high temperature, drought, and high salt conditions.
MATERIALS AND METHODS
Study Region, Samples Collection and RNA Extraction
The site chosen for studying was a Rhazya stricta community and public site (Hadda, Mecca-Jeddah road). The sampling site was assigned coordinates via GPS for ease of relocation (N21° 45ʹ 04.03", E39° 53ʹ 88.92"). The leaves were taken from the five plants (Rhazya stricta, Enneapogon desvauxii, Citrullus colocynthis, Senna Italica, & Zygophyllum simplex). The samples were packaged in plastic bags contained 5 aluminum foil envelops and stored at -80ºC until analysis. After tissue collection, each pooled sample was ground in liquid nitrogen. Total RNA was extracted in Trizol (Life Technologies, NY, USA) according to manufacturer instructions.
Sequencing and Data Processing
RNA-seq libraries were prepared by BGI genomics by TruSeq library creation protocol (Illumina, Hog Kong, China). Samples were sequenced using the Illumina HiSeq-2000 platform.
Assembly and Annotation
Firstly, we filter low-quality, adaptor-polluted and high content of unknown base (N) read to get clean reads. And then perform the de novo assembly with clean reads to get the Unigenes, after that, Unigene expression analysis, Unigene functional annotation are performed.
Unigene Functional Annotation
NT, NR, GO, COG, KEGG, SwissProt, and InterPro are functional databases (for more details, please find the database below). We use Blast align Unigenes to NT, NR, COG, KEGG, and SwissProt to get the annotation, use Blast2GO with NR annotation to get the GO annotation and use InterProScan5 to get the InterPro annotation. Software Information:
Unigene CDS Prediction
With functional annotation, we select the segment of Unigene that best mapped to functional databases in priority order of NR, SwissProt, KEGG, COG as its CDS, and display from 5' to 3' in FASTA format. Unigenes that can't be aligned to any database mentioned above are predicted by ESTScan with Blast-predicted CDS as a model.
Unigene Expression
We mapped clean reads to Unigenes using Bowtie2 and then calculated gene expression level with RSEM.
Identification of Orthologous between Five Samples
The five different plants grew together in the same environment and subjected to the same stresses; there should be some common genes’ influence may be the common orthologous genes’ expression. The orthologous genes were detected and then analyzed together. For example, Blast software was used to align the unigenes between Rhazya stricta and other four samples, then Filter the reliable alignment genes (filter condition % identity >30, e-value < 1e-10, bit score >200, alignment length >60%), based on the expressed genes (common gene expressed in five samples) and the filtered genes we get the orthologous genes and extract annotation (NR, NT, Swissport, KEGG, COG, Interpro, GO). Finally, depending on the annotation we highlight the genes about the stresses.
Unigene TF Prediction
The getorf was used to find ORF of each Unigene, then align ORF to TF domains (form PlntfDB) use hmmsearch, and identify TF according to the regulations described form PlantfDB. Based on the annotated TF Family, the common annotated TF domains in Five samples were selected.
RESULTS
Principal component analysis (PCA) for these plant species based on their profiles of TF-families (with the ratios of 59 TF families as variables and the 5 plants as samples; Fig 1) revealed that the TF-family profiles of the five lineages (Rhazya stricta, Enneapogon desvauxii, Citrullus colocynthis, Senna Italica, and Zygophyllum simplex) are all quite distinct from each other, whereas those of the species within each of the lineage are more similar. The three plant lineages (E. desvauxii, C. colocynthis, and Z. simplex) can be separated on PC1 level (accounting for 65.93% of cumulative variance), while R. stricta and S. Italica can be distinguished from each other on PC2 level (accounting for 23.6% of cumulative variance). Therefore, these TF-families appear to play particularly prominent roles within higher plants.
Venn diagram analysis identified unique 225 transcripts in each of R. stricta and E. desvauxii, 85 transcripts in C. colocynthis, 92 transcripts in S. Italica, and 41 transcripts in Z. simplex, respectively. R. stricta shared TFs with E. desvauxii and Z. simplex in 225 which is the highest number, while with C. colocynthis in 85, and S. Italica in 92 transcripts. The lowest sharable TFs were found between S. Italica and C. colocynthis in 26 transcripts only. All five plants were shared in only 15 transcripts (Fig 2).
A total of 225 transcripts were detected in the five plants. R. stricata was the highest plant in upregulated transcripts with 171, followed by E. desvauxii with 124 transcripts, S. Italica with 50 transcripts, and C. colocynthis and Z. simplex with 30 transcripts for each (Fig 3).
We further characterized genes that have significant gene ontology (GO) terms (≤ 0.05) in biological processes, cellular components, and molecular functions with 1420 genes (450 genes in R. stricta, 475 genes in E. desvauxii, 197 genes in C. colocynthis, 223 genes in S. Italica, and 75 genes in Z. simplex) were significantly assigned with GO terms for 29 transcriptional processes. Among the GO terms associated with response to stimuli in biological processes, the most significant categories were a response to cold, light, abscisic acid, water deprivation, oxidative stress, salt stress, and heat stress (Fig 4).
The most abundant transcription factor families in the five plants were MYB (1105 transcripts), MYB-related (803 transcripts), bHLH (536 transcripts), and AP2-EREBP (472 transcripts). The highest number of TF family was MYB with 288 transcripts in Z. simplex, flowed by C. colocynthis with 221 transcripts. NOZZLE transcription factor family was not transcribed in E. desvauxii, while the LFY TF family was transcribed only in C. colocynthis. The zinc TF families were the most transcribed and represented by 8 TF families (PLATZ, C2C2-CO-like, C3H, VOZ, C2C2-GATA, C2C2-DOF, C2H2, and ZF-HD), with the highest one (C3H TF family, 99 transcripts in both R. stricta and Z. simplex. The highest number of TF families was transcribed in Z. simplex (2195 TF family) followed by C. colocynthis (1713 TF family) (Figs 5A-E).
The regulatory network showed that some genes involved in the heat response in the five plants under this study, 12 TF genes were upregulated in R. stricta and controlled several genes in some vital processes inside the plant, while E. desvauxii and S. Italica respond to heat by upregulation of 6 TF genes for each, C. colocynthis by regulation of 3 TF genes and the least one was Z. simplex by upregulation of two TF genes only. The most common upregulated TF genes was SHMT (Serine hydroxymethyl transferase) which involved in PATHWAY: One-carbon metabolism; tetrahydrofolate interconversion (Fig S1, Fig 6) found in R. stricta, E. desvauxii, C. colocynthis, and S. Italica, followed by LHCA1 (Light-harvesting complex) involved in photosystem I (Fig S2) found in R. stricta, E. desvauxii and S. Italica, CAB21 (Chlorophyll a-b binding protein 21) involved in photosystem II (Fig S2), and also CAB151 (Chlorophyll a-b binding protein 151) involved in photosystem II (Fig S2). The least plant had fewer genes in response to heat was Z. simplex by upregulation of two TF genes FTSH2 (ATP-dependent zinc metalloprotease) involved in photoinhibition and LHCA-P4 (Chlorophyll a-b binding protein P4) involved in photosystem II (Fig S2, Fig 6).
On the contrary, the most common TF gene was GOLS2 (Galactinol synthase 2) (EC 2.4.1.123) involved in galactose metabolic process and found in all plants except Z. simplex (Fig 7, Fig S3), followed by Histone H4, which is one of the five main histone proteins involved in the structure of chromatin in eukaryotic cells and found in R. stricta, E. desvauxii, and Z. simplex and finally, ACO3 (Aconitate hydratase 3) involved in tricarboxylic acid cycle pathway (Fig 7), also AB1K1 (Non-specific serine/threonine-protein kinase) (EC 2.7.11.1). This is a heterogeneous group of serine/threonine protein kinases that do not have an activating compound and are either non-specific or their specificity has not been analyzed to date (Fig S4), both were found in R. stricta and E. desvauxii.
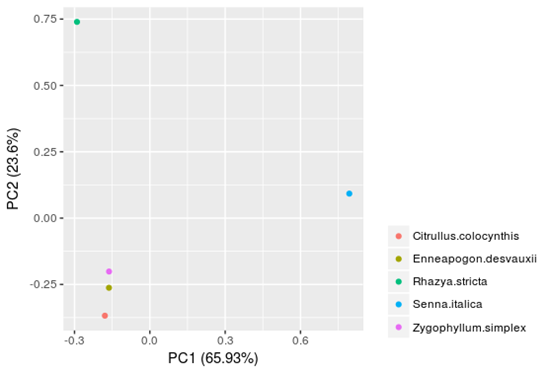
Figure 1. Principal component analysis (PCA) for the five plants (Rhazya stricta, Enneapogon desvauxii, Citrullus colocynthis, Senna Italica, & Zygophyllum simplex) was performed based on TF-family profiles. (A) Two-dimensional PCA results based on PCA1 and PCA2. (B) Three dimensional PCA results based on PCA1, PCA2, and PCA3.
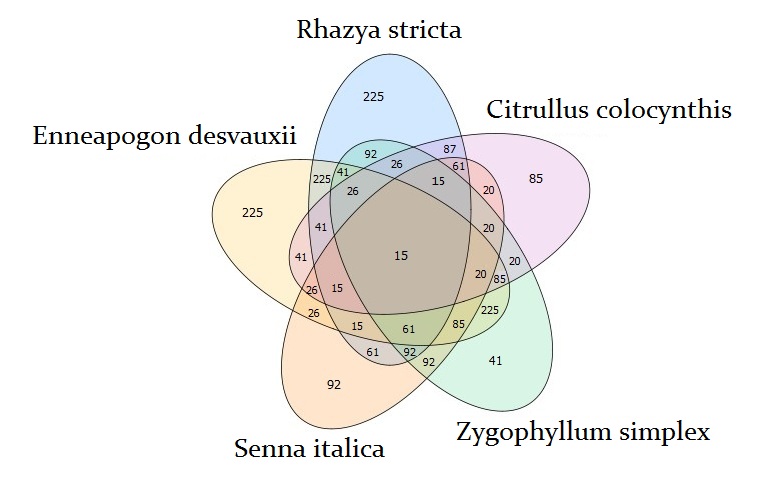
Figure 2. Venn diagram shows the overlap among transcription factors that were found to be significantly upregulated for each of the indicated comparisons between the five plants (Rhazya stricta, Enneapogon desvauxii, Citrullus colocynthis, Senna Italica, and & Zygophyllum simplex).
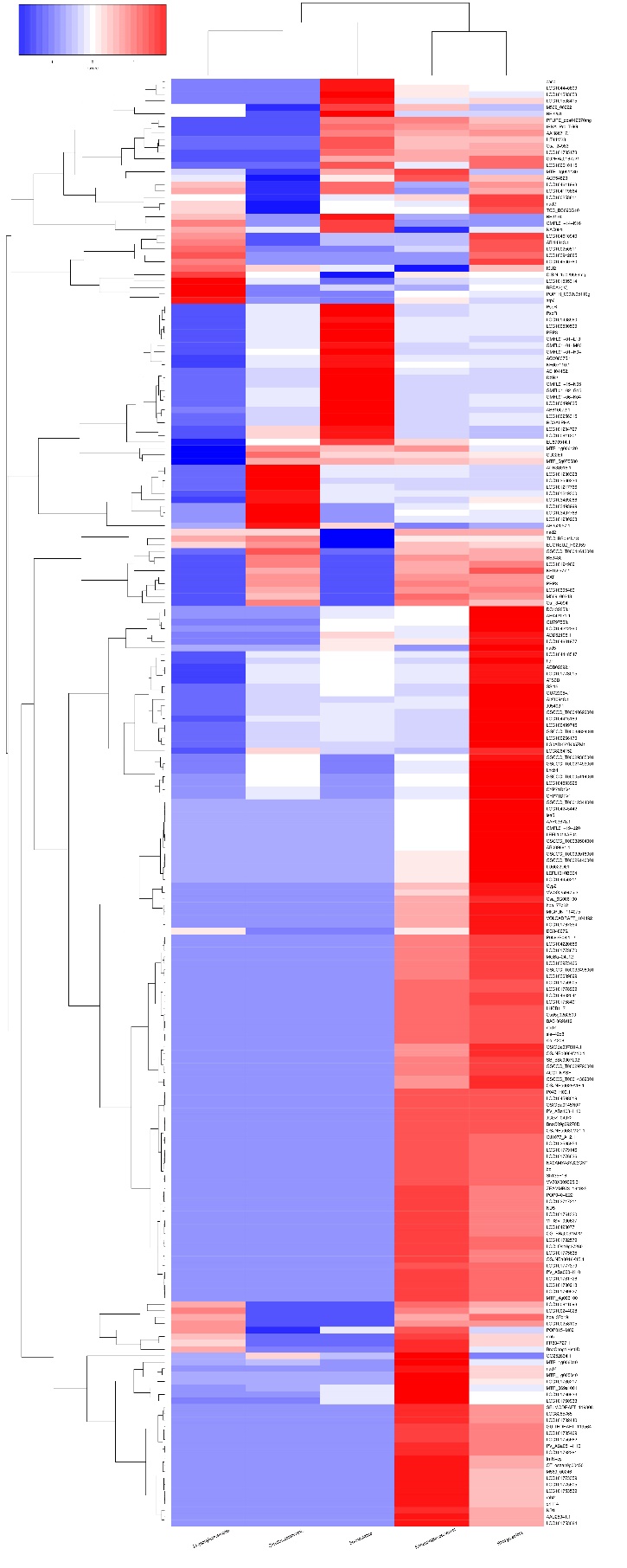
Figure 3. Cluster analyses of differentially expressed genes (DEGs) among the five plants (Rhazya stricta, Enneapogon desvauxii, Citrullus colocynthis, Senna Italica, & Zygophyllum simplex). The color key represents the value of log10 (RPKM). Red represents highly expressed (up-regulated) genes, while blue represents down-regulated genes.
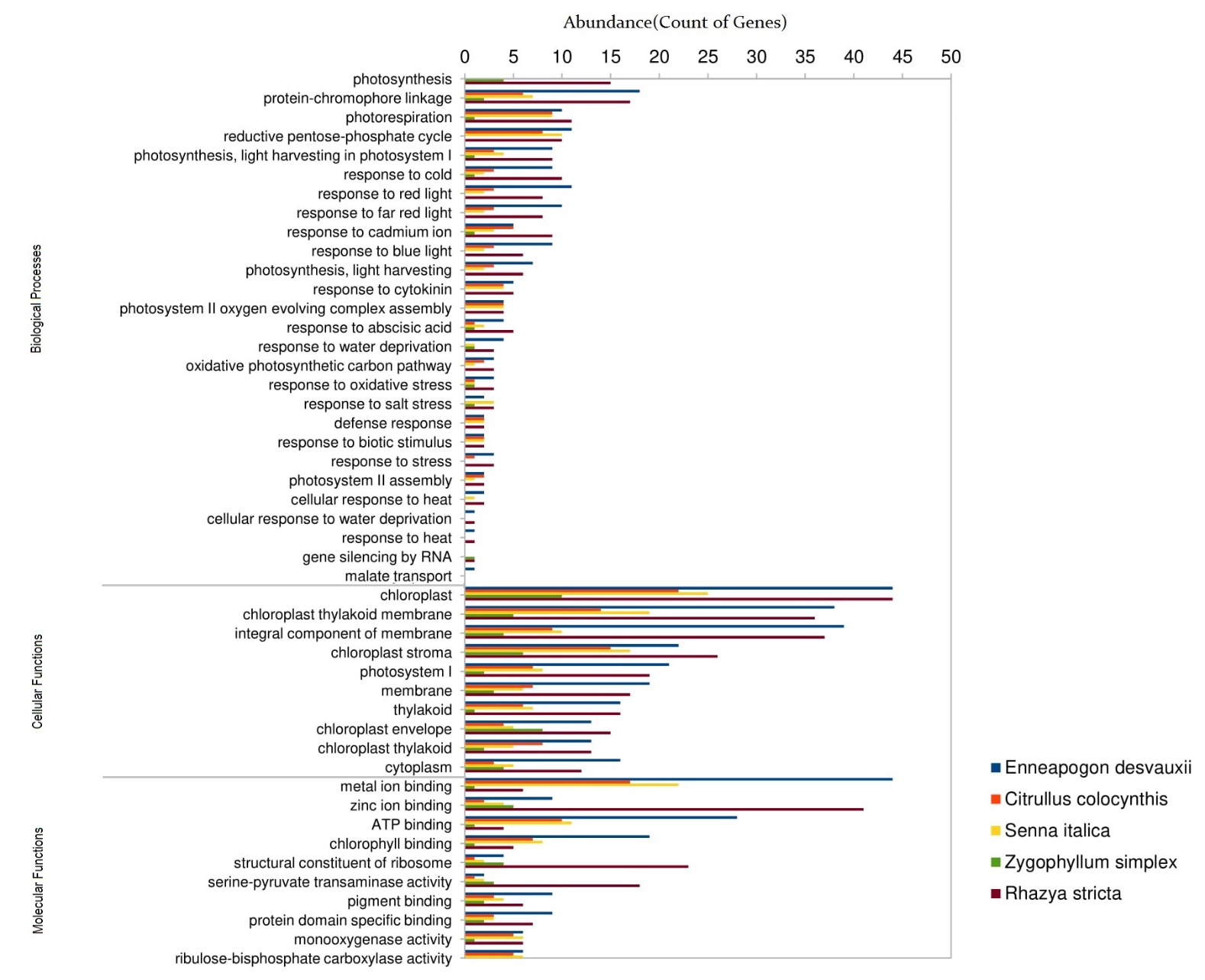
Figure 4. Analysis of Gene Ontology (GO) Enrichment for Differentially Expressed Genes (DEGs)
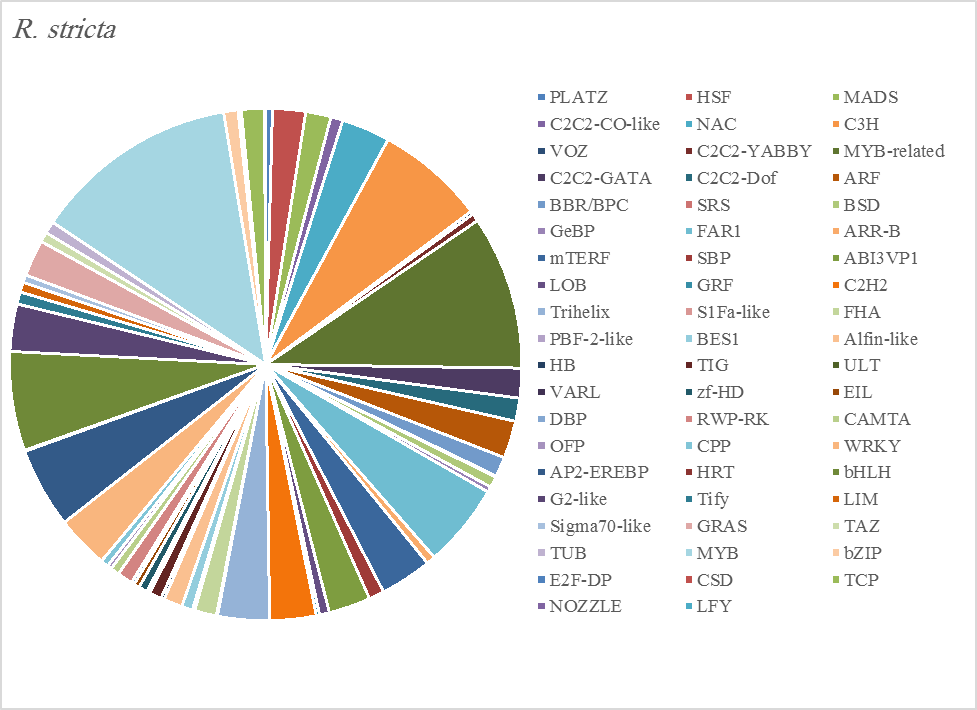
Figure 5A. Distribution of Differentially Expressed Transcription Factors in Gene Families for Rhazya stricta
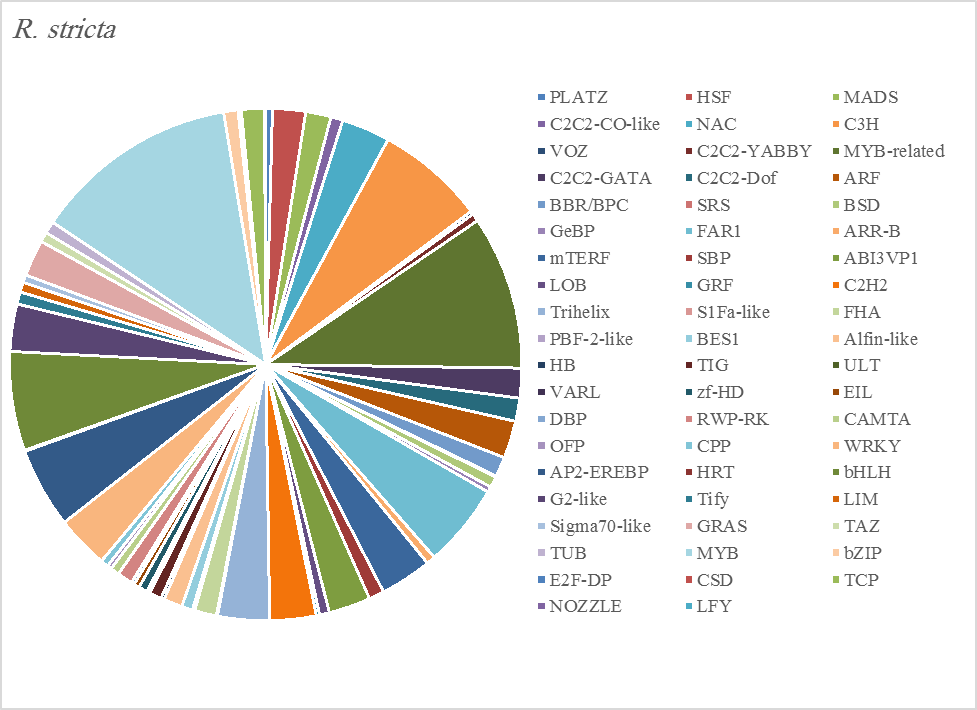
Figure 5B. Distribution of Differentially Expressed Transcription Factors in Gene Families for Enneapogon desvauxii
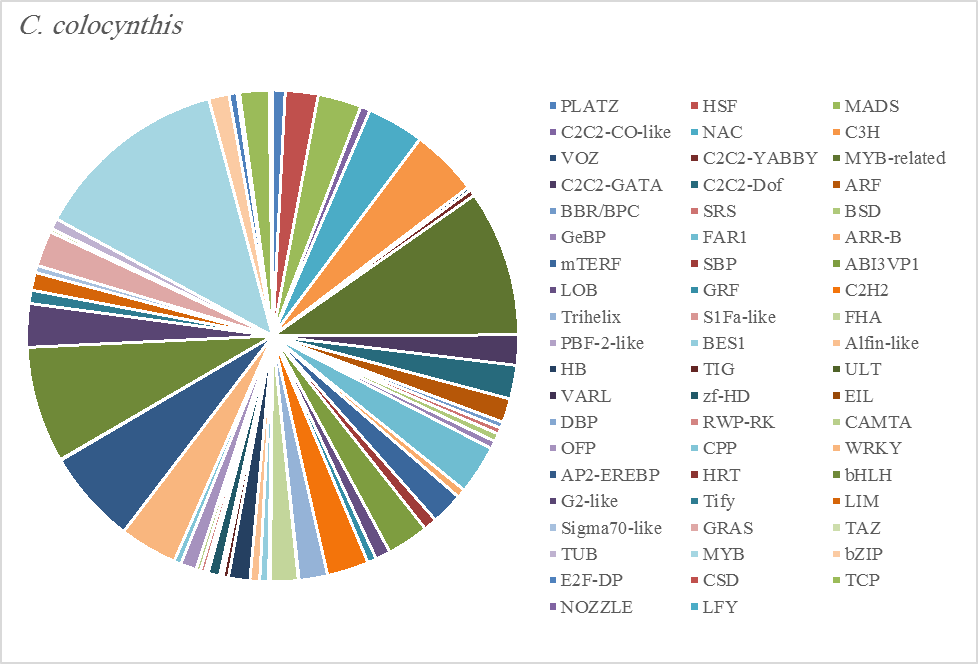
Figure 5C. Distribution of Differentially Expressed Transcription Factors in Gene Families for Citrullus colocynthis
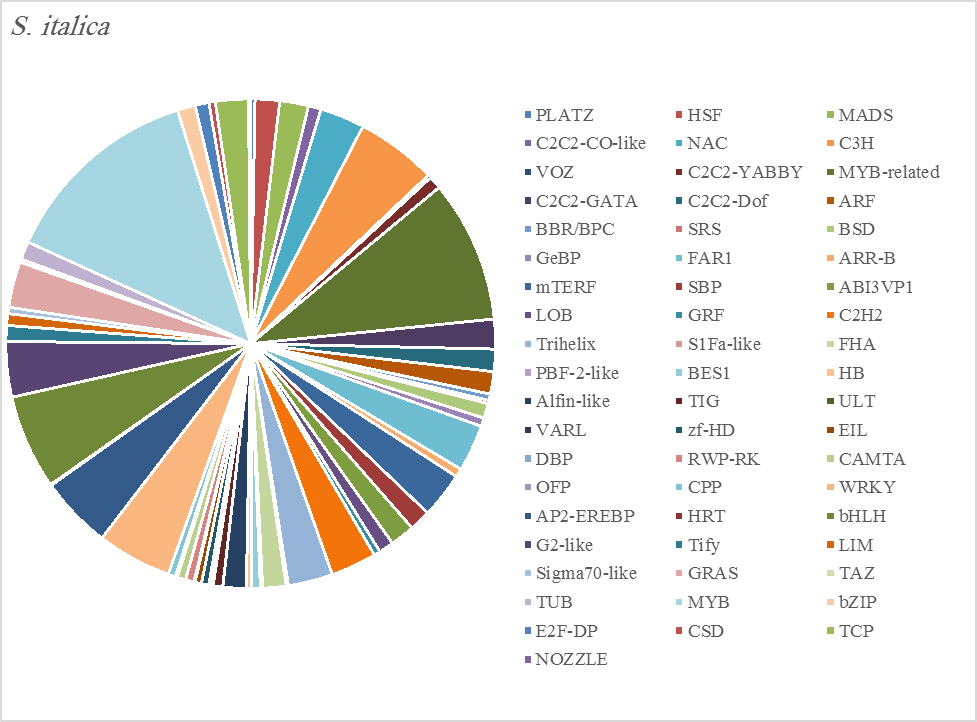
Figure 5D. Distribution of Differentially Expressed Transcription Factors in Gene Families for Senna Italica
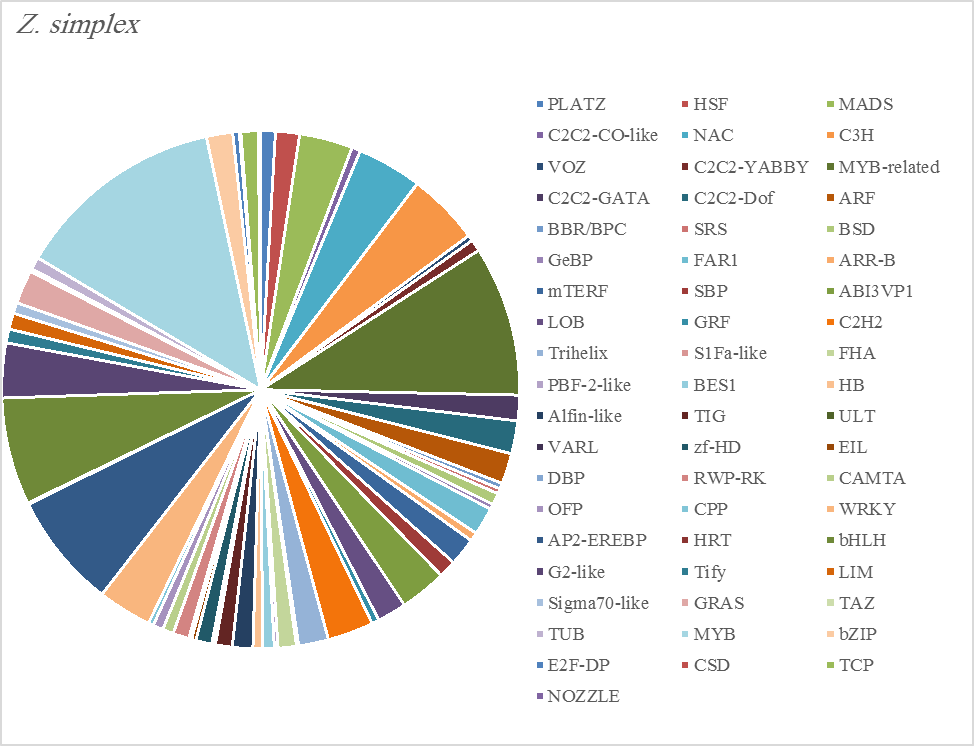
Figure 5E. Distribution of Differentially Expressed Transcription Factors in Gene Families for Zygophyllum Simplex
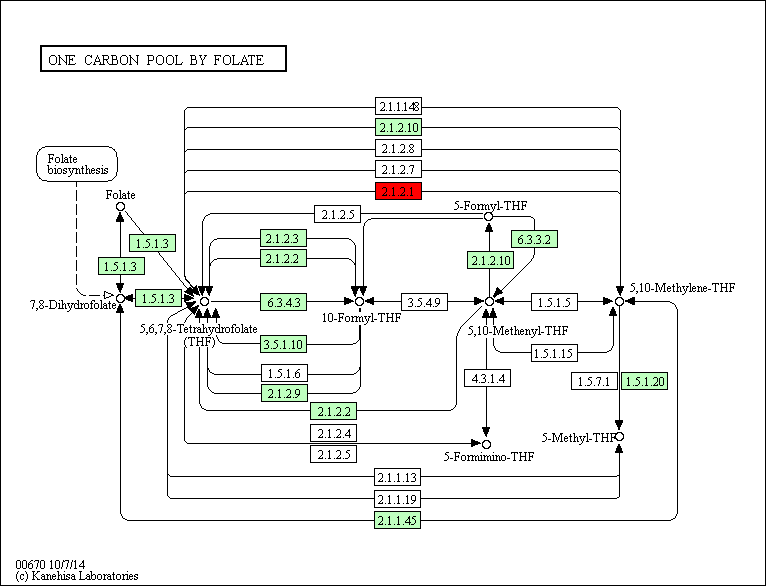
Figure S1: One-carbon Metabolism Involved in Photosystem I, II, and III
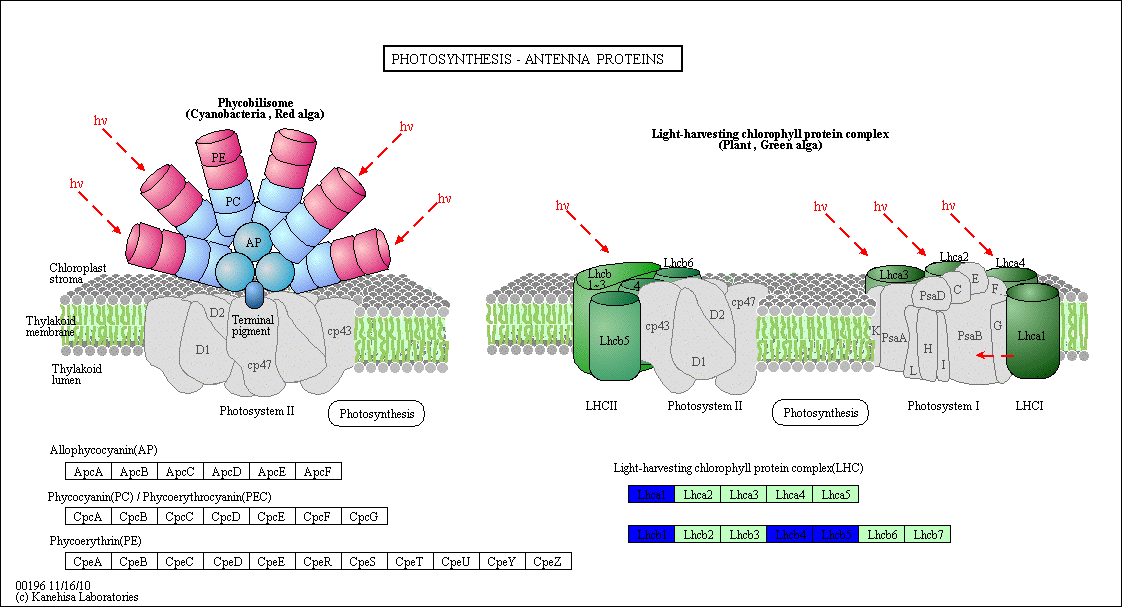
Figure S2: Photosynthesis- Antenna Protein Structure
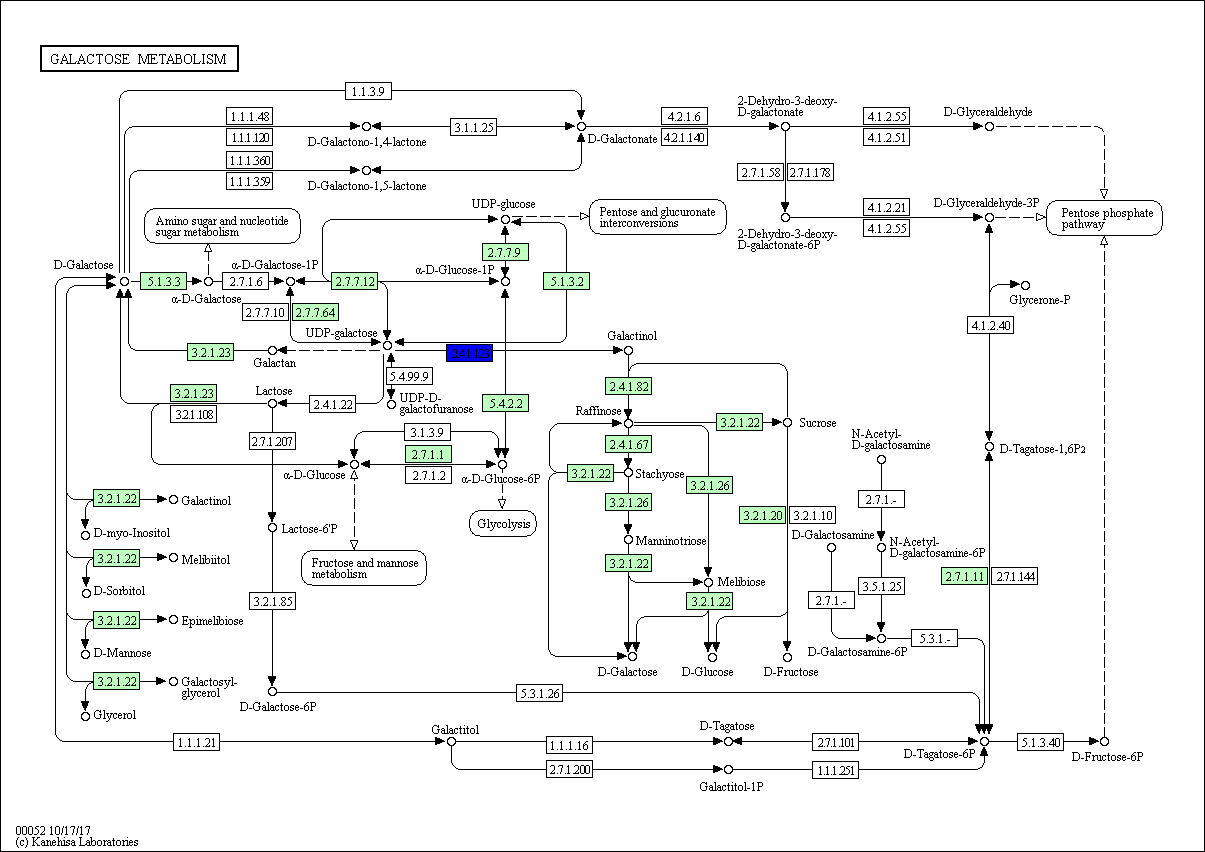
Figure S3: Galactose Metabolic Process
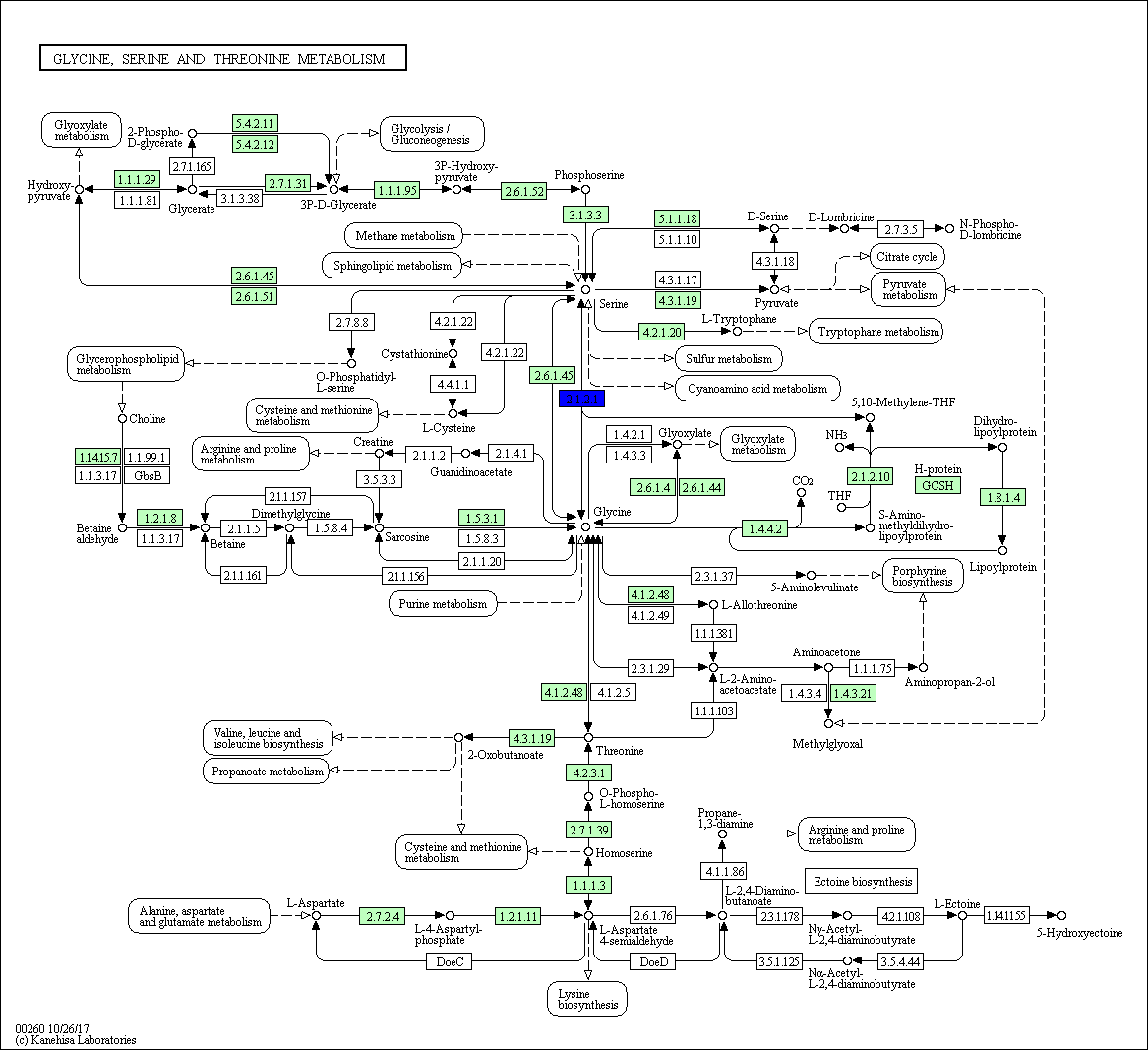
Figure S4: Glycine, Serine, and Threonine Metabolism Pathway
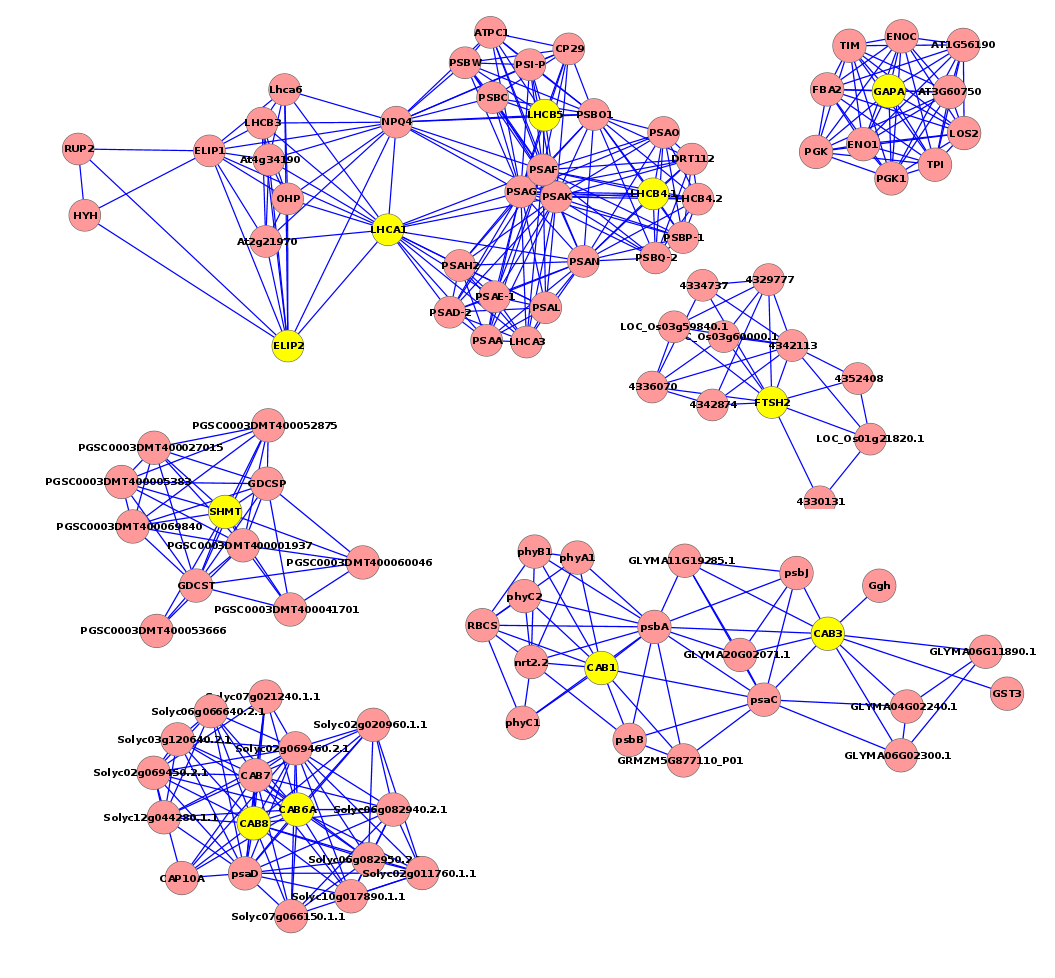
Fig 6. This regulatory network shows some genes involved in the heat response in the five plants (Rhazya stricta, Enneapogon desvauxii, Citrullus colocynthis, Senna Italica, & Zygophyllum simplex), each “yellow dot” represents a TF gene and each “lavender dot” represents a target gene.
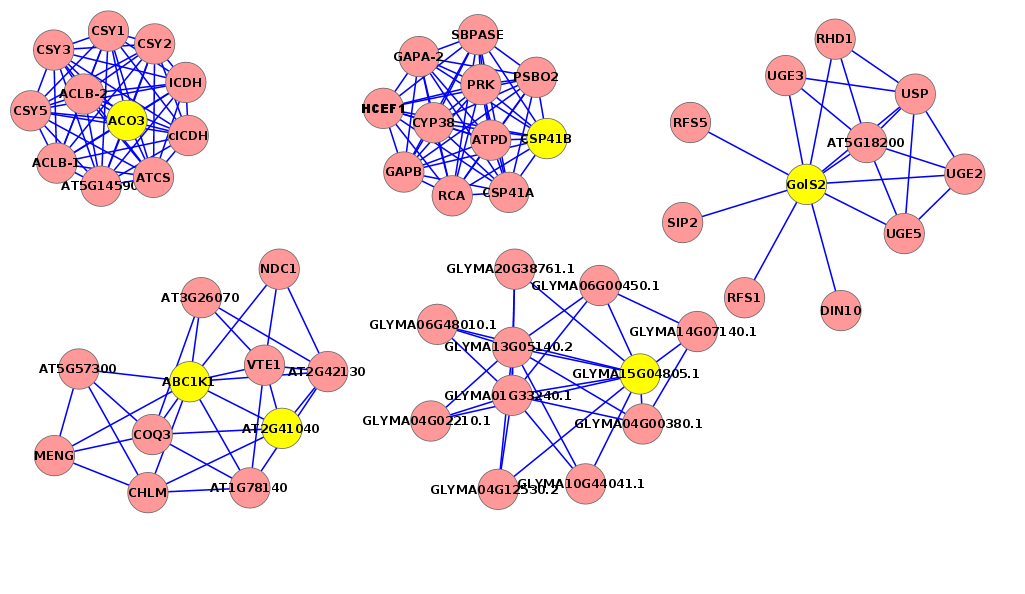
Figure 7. This regulatory network shows some genes involved in the salt response in the five plants (Rhazya stricta, Enneapogon desvauxii, Citrullus colocynthis, Senna Italica, & Zygophyllum simplex), each “yellow dot” represents a TF gene and each “lavender dot” represents a target gene.
DISCUSSION
The regulatory elements of the recognized stress genes induced were studied, including abscisic acid (ABA), dehydration response element (DRE), heat shock elements (HSEs), and responsive promoter elements (ABREs). These elements give the binding site of transcription factors families (TFs) to grant expression of the stress-responsive gene [10-12].
The principal component analysis (PCA) could be used to expose patterns, exclude repetition in data sets and reduce relatively a large series of data into a smaller number of components by looking for groups that have very strong inter-correlation in a set of variables and each component explained percent (%) variation to the total variability [13] as morphological and physiological diversity frequently appear in crop species. The first principal component is the largest contributor to the total variation in the population followed by subsequent components. The first three principal components are usually the most critical in reverse the variation of the patterns among accessions, and the biomolecule reports [14]. In this regard, our study demonstrated that the TF-family profiles of the five lineages (R. stricta, E. desvauxii, C. colocynthis, S. Italica, & Z. simplex) are all noticeable from each other. The three plant lineages (E. desvauxii, C. colocynthis & Z. simplex) can be separated on the PC1 level (65.93%), while R. stricta and S. Italica can be distinguished from each other on the PC2 level (23.6%).
4982 transcripts are overlapping as light-stress responses between the systemic and local leaves in Arabidopsis. 5363 transcripts in systemic leaves and 6502 transcripts in local leaves of rbohD plants were significantly upregulated as a result of the light stress response [15]. Our findings agreed with these results, where all the five plants were shared in only 15 transcripts. The highest plant in upregulated transcripts in response to abiotic factors was R. stricta (171), followed by E. desvauxii (124), S. Italica (50), and C. colocynthis and Z. simplex (30 each).
It has been suggested that modifications in the genes encoding TF expression function as one of the main exporters that regulate the evolution of higher plants; moreover, the possible link between the evolution of plant and profiles of TF-family is still difficult to find. In the Gene Ontology (GO) analysis, 52 GO-slims and GO terms enriched in each predicted TFBS motif of target genes were identified to detect the TFBS motif main function in Nannochloropsis. For the most conserved 68 TFBSs motifs, 40 on the biological process (BP) level and 36 on molecular function (MF) level was found enriched [16]. In this regard, our findings concluded that the enriched GO terms for R. stricta (450), E. desvauxii (475), C. colocynthis (197), S. Italica (223), and Z. simplex (75) were significantly identified and assigned to the main functions of transcription factor binding site (TFBS) motif predicted. In R. stricta 158 GO-terms on BP level, 69 on cellular components (CC) level and 122 on MF level, in E. desvauxii 157 on BP, 67 on CC and 119 MF levels, in C. colocynthis 50 on BP, 38 CC and 58 on MF levels, in S. Italica 72 on BP, 43 on CC and 63 on MF levels, and in Z. simplex 70 on BP, 39 on CC and 43 on MF levels.
It was found that MYB, WRKY, NAC, bHLH, bZIP, HD-zip via ABA-independent or -dependent pathways play a crucial role in tolerance of drought [17, 18]. The proteins expression that contains specific domains of DNA-binding like bZIP/HD-ZIP, AP2/ERF, MYB, NAC, MYC, and WRKY may be induced or repressed under many stress conditions in Arabidopsis [10, 19-23], while in maize, a set of 10 TF families (ERF, AP2, MYB, bHLH, bZIP, GRAS, NAC, WRKY, NF-YA & NF-YB) were identified to regulate diverse molecular and physiological functions such as hormone signaling, stomatal regulation, osmoregulation, and root development [24, 25]. 82 bHLH genes was found in wheat and had orthologs 27 and 28 TFs in Arabidopsis and rice, and 27 TFs in both [26]. On the other hand, there are similar roles of AtNF-YB1 in Arabidopsis and ZmNF-YB2 in maize in enhancing the performance of crops under drought stress [27]. bZIP TF has a fundamental role in plant growth, ABA signaling, and abiotic stresses. In the maize seedling stage, ZmbZIP72 is one of bZIP TF which over-expressed in response to salinity, drought, and ABA stress in different organs [18]. Considering this, our study revealed that the most abundant TF families in the five plants were MYB followed by, MYB-related, bHLH, and AP2-EREBP (1105, 803, 53, & 472 transcripts, respectively). The elevated number of TF family was MYB (288) in Z. simplex, followed by C. colocynthis (221). NOZZLE TF family was not transcribed in E. desvauxii, while the LFY TF family was transcribed only in C. colocynthis.
Several zinc finger domains class expression may be induced or repressed under many stress conditions in Arabidopsis [19-22]. In rice, a zinc finger-containing protein C2H2-type (DST & SNAC1) were engaged in the opening/closure of stomata [28, 29] and play a fundamental role in the ROS detoxification and osmoprotectants synthesis [30]. In conclusion, sequence motifs repeat like zinc fingers C2H2 are accountable for the accelerated expansions of TF families [31]. Our findings agreed with these studies where zinc finger TF families were the most transcribed families and represented by 8 TF families (PLATZ, C2C2-CO-like, C3H, VOZ, C2C2-GATA, C2C2-DOF, C2H2 & ZF-HD), with elevated one (C3H TF family, 99 transcripts in both R. stricta & Z. simplex).
Heat stress tolerance is a multigenic process with many regulatory mechanisms acting during plant development [32, 33]. During sexual reproduction, heat stress response and damage are more visible in plant leaves [34] and pollen [35] than other tissues. Heat shock proteins (HSPs) are proteins family that are produced in response to stress conditions exposure by cells. Photosynthesis is highly sensitive to heat and might be suppressed by heat stress conditions. Modification in the processes of photosynthesis, including photosystem I and II (PS I, PSII), electron transport chains, synthesis of ATP, and fixation of carbon regularly occur when plants are exposed to heat stress [36-38]. The adjustment in Photosynthesis in response to heat stress has been well reported in the past few years [36-43]. The effect of heat stress on the photosynthesis process is shown in different levels of the plants including molecular, physiological, and biochemical aspects, such as the thylakoid membrane destacking, damage in photosystem II, cytochrome b6/f complex suppression, and ribulose-1, 5-bisphosphate carboxylase/oxygenase (RuBisCO), in addition to that the production of reactive oxygen species (ROS) [36]. HSP 70 overexpression could increase drought and salinity tolerance [44, 45]. WHIRLY1 interacts with LHCA1 and affects the genes encoding LHCI and photosystem I (PSI) and expression [46]. The thylakoid FtsH protease complex consists of FtsH1/FtsH5 (type A) and FtsH2/FtsH8 (type B) subunits in Arabidopsis [47]. Our findings agreed with that results, the regulatory network revealed that as heat response, 12 genes were upregulated in R. stricta and controlled various genes control essential processes inside the plant, while E. desvauxii and S. Italica respond to heat by upregulation of 6 genes for each, C. colocynthis by regulation of 3 genes and the least one was Z. simplex by upregulation of two genes only. The most common upregulated TF genes were SHMT found in R. stricta, E. desvauxii, C. colocynthis, and S. Italica, followed by LHCA1, CAB21, and CAB151 found in R. stricta, E. desvauxii, and S. Italica. The fewer genes in response to heat were in Z. simplex by upregulation of two TF genes FTSH2 and LHCA-P4, this may be attributed to its nature where it stores water in its all parts. On the contrary, the most common TF gene was GOLS2 found in all plants except Z. simplex, followed by Histone H4 found in R. stricta, E. desvauxii, and Z. simplex and finally, ACO3, also AB1K1, both were found in R. stricta and E. desvauxii.
CONCLUSION
The regulatory network in desert plants showed that a number of genes involved in the environmental stress response in the five tested medicinal plants. Some genes were upregulated and controlled several vital genes in some plants, while other plants responded to the environmental stress by upregulation of other genes. All these responses contribute to highlight the stress tolerance of these plants. Biochemically, the plants modify different metabolism pathways to adapt to this environmental stress especially in a stressful environment like the Saudi desert.
ACKNOWLEDGEMENT
This project was funded by the Deanship of Scientific Research (DSR) at King Abdulaziz University, Jeddah, under grant no. (HiCi 1-363-1434H). The authors, therefore, acknowledge DSR for technical and financial support.
REFERENCES


